Like any other regulated product, your medical device must be properly labeled. But do you know how to prepare a medical device label and which regulations your label has to comply with? And how about which ISO standards can help you meet these regulatory requirements? Let’s look into that! We’ll start with how labeling is defined by the US FDA and EU MDR regulations.
What Is Medical Device Labeling?
According to the FDA, medical device labels need to meet labeling requirements. When a medical device has not complied with these requirements, the FDA takes action according to the primary law of the Federal Food, Drug, and Cosmetic Act (FFD & C Act).
Part 801 of 21 CFR refers to Section 201(k) of the FFD & C Act for the definition of what constitute as a label:
“[…] a display of written, printed, or graphic matter upon the immediate container of any article; and a requirement made by or under the authority of this Act that any word, statement, or other information appear on the label shall not be considered to be complied with unless such word, statement, or other information also appears on the outside container or wrapper, if any there be, of the retail package of such article, or is easily legible through the outside container or wrapper.”
Section 201(m) then defines labeling as:
“all labels and other written, printed, or graphic matter
1) upon any article or any of its containers or wrappers, or
2) accompanying such article.”
Similarly, EU MDR 2017 defines label in a European context as:
“A printed, written, or graphic information that appears either on the device itself or on the packaging of a unit or on the packaging of multiple devices.”
Why Is Medical Device Labeling so Important?
The end user of a medical device could be a patient, medical professional, or otherwise experienced user. All end users need instructions, guidance, and warnings to properly use a medical device safely. This requires that all potential risks were thought of and eliminated by the manufacturer, and required warnings and instructions were added to the labels. Risks may cause just a mild error – or serious injury or death. Regardless of the level of risk, any issue will have a negative impact on the patient, even if it is just a minor problem. If you thoroughly read and apply labeling requirements, update your label according to the cases you have received from users. This will decrease the cost of a bigger problem, eliminate possible complaints, and – most importantly – keep your patients safe.
Requirements for Medical Device Labeling
When you plan to market a medical device in the US, you first need to prepare a proper label for each product. The requirements for medical device labeling are extensive; elements can vary depending on different factors, such as the type or design of the medical device. Ultimately, it is the responsibility of the manufacturer to ensure that all necessary elements are covered and that the label successfully conveys all essential information to address the patient’s needs.

Do you have the full list of regulatory documents on labeling requirements?
Check this downloadable for an easy overview.
Regulatory Requirements by Authority
The best way to review and approve labeling content is to create a labeling checklist for each regulatory requirement that apply to your device. You can use the following tips to create a checklist:
- List regulatory authorities that you need to comply with.
- List all general requirements for labeling, direction for use, and instruction for use.
- Think about the labeling needs that are specific to your device.
- Review the ISO label standards that are specific to your device.
- Include necessary warnings about all known risks, including dangers of off-label use.
- Include the required symbols that you need on your label.
- Last but not least, engage your labeling process with usability testing to see if your label has been designed well.
1. Labeling Requirements in the United States
The labeling of in vitro diagnostic products and medical devices is governed by two federal laws for the US:
-
- The Fair Packaging and Labeling Act (FPLA)
- The Federal Food, Drug, and Cosmetic Act (FFD & C Act)
Most of the provisions of the FPLA and the FFD & C Act are codified in the following parts of Title 21 of the US Code of Federal Regulations (CFR):
21 CFR Part 801 General Device Labeling
Subpart 801.15 explains the use of symbols in medical device labeling. Additionally, FDA issued the Use of Symbols in Labeling under the Final Rule in June 2016. This rule explicitly allows graphical representations of information to be included on the label of medical devices and certain biological products. Symbols are allowed on labels without explanatory text if specific requirements are met. FDA also revised prescription device labeling regulations to allow the use of the symbol statement “Rx only” or “℞ only” in the labeling.
Medical device manufacturers should have a glossary of symbols. The labeling should explicitly show the end-user where to find that glossary.
The following elements should be included on an FDA-compliant label:
-
-
- Name and address of the manufacturer
- Intended use of the device
- Adequate instructions for a layperson to safely operate the device
- No false or misleading statements
- Must be displayed prominently in an appropriate location
- Unique Device Identifier (UDI)
-
21 CFR Part 809 In Vitro Diagnostic Products (IVD)
This includes reagents, instruments, and systems that are used to diagnose a disease or condition, or to determine the state of health to cure, mitigate, treat, or prevent disease. These products are intended for use in the collection, preparation, and examination of specimens taken from the human body.
In Subpart 809.10, labeling for in vitro diagnostic products is explained: The label for an IVD should state the listed information (where applicable).
21 CFR Part 812 Investigational Device Exemptions (IDE)
This part provides procedures for conducting clinical investigations of devices. The approved investigational device exemption (IDE) permits a device that otherwise would have to comply with a performance standard or receive premarket approval to be shipped lawfully for research purposes.
All IDEs (Investigational Device Exemptions) which are approved or considered approved, exempt a device from the following requirements of the FFD & C Act and regulations issued thereunder:
-
-
- Misbranding
- Registration, listing, and premarket notification
- Performance standards
- Premarket approval
- Banned device regulation
- Records and reports
- Restricted device requirements
- Good Manufacturing Practice (GMP)
- Color additive requirements
-
Additionally, the label should state: "CAUTION - Investigational device. Limited by Federal [or the United States] law to investigational use."
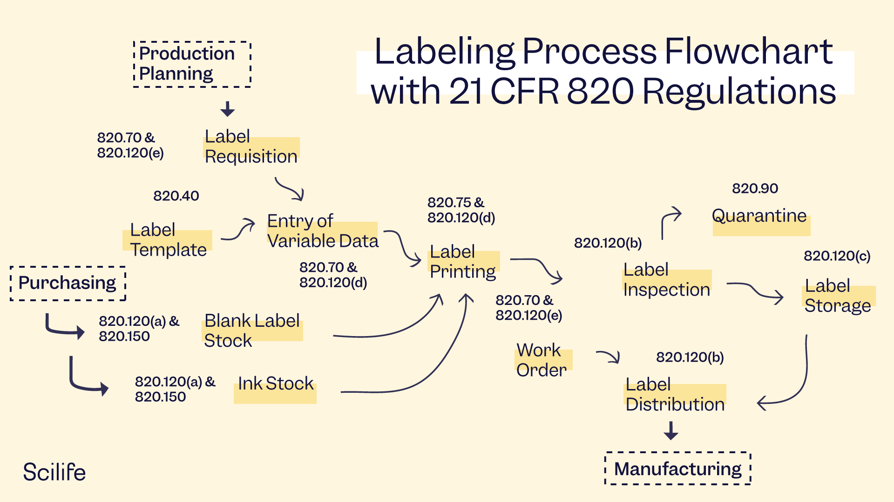
21 CFR Part 820 Good Manufacturing Practices (GMPs)
Medical device labeling requirements are outlined under Subchapter K on labeling and packaging control, in Subpart 820.120 on device labeling. This section requires manufacturers to establish a proper labeling process and procedures to control labeling activities such as label integrity, labeling inspection, storage, operations, and control number. Additionally, all documentation should be part of your Quality System.
21 CFR Part 830 Unique Device Identification (UDI)
All medical device packages should bear a Unique Device Identifier (UDI) that meets the requirements of Subpart 801.20 and Part 830. The requirements for a UDI should include the following elements:
-
-
- Issued under a system operated by the FDA or an agency accredited by the FDA.
- Ensure compliance with the following international standards:
- ISO/IEC 15459-2
- ISO/IEC 15459-4
- ISO/IEC 15459-6
- Only ISO/IEC 646 invariant characters and numbers are used.
- Include the Unique Device Identifier (UDI) on labels and packages.
- Submit your medical device to the Global Unique Device Identification Database (GUDID).
-
21 CFR Part 1010 General Electronic Products
Performance standards for electronic products are explained under Part 1010. Labeling requirements are specifically outlined under 21 CFR Part 1010.3 Identification. Among other labeling requirements, you should include information on the place, month, and year of manufacture.
2. Labeling Requirements in Europe
If you plan to market your medical devices in Europe, your products must meet the labeling requirements under the European Medical Devices Regulation (EU MDR). This relatively new regulation caused the number of labeling requirements to increase when compared to the previous EU Medical Devices Directive (MDD), which brings many challenges for medical device manufacturers.
Medical device manufacturers need to spend more time meeting the requirements of the EU MDR as device safety and clinical effectiveness data should be transparently shared with end users. The EU MDR describes medical device safety and performance requirements in Annex I on general safety and performance requirements.
Further reading: EU MDR: Key Changes and Important Steps
Label and Instruction for Use (IFU) requirements are outlined here in the EU MDR:
Annex I. General Safety And Performance Requirements
Chapter III. The Requirements Regarding the Information Supplied with The Device
10.4.5. Labeling
The label should bear the following details:
-
- The device’s purpose must be clearly stated on both the label and in the instructions. This information enables users to identify the device or other contents and any operating instructions, warnings, or precautions to take.
- Each medical device must be accompanied by as much information as is necessary for users. Manufacturers should consider the training and knowledge needs of the users.
- The basic instructions should appear on the label with a more detailed copy to be included in the enclosed instructions.
- The instructions must contain:
- All details required on the label,
- Any side effects from the use of the device, and
- Details for its correct use, including any specific precautions.
- The required information should be made available to the user and the patient in their national language(s) or in another community language when a device reaches the final user.
- Manufacturer’s contact details:
- The name of the medical device manufacturer, the registered trade name, or the registered trademark, and
- The address of the manufacturer's place of business.
- Contact details (name and address) of the authorized representative if the manufacturer is based outside of the EU.
- If applicable, there should be an indication that the device contains or incorporates a medicinal substance, including:
- Cells or tissues of human origin or their derivatives,
- Cells or tissues of animal origin or their derivatives, and
- Plasma or human blood derivative.
- Where devices, parts, or materials used therein contain substances, the label should include the following information:
- Presence of mutagenic, carcinogenic, or toxic for reproduction,
- Presence of endocrine-disrupting substance,
- Presence of a medicinal substance or tissue/cells,
- Absorption rate,
- Blood or tissue derivate, and
- Innovations like nanotechnology or computer software.
- The device’s purpose must be clearly stated on both the label and in the instructions. This information enables users to identify the device or other contents and any operating instructions, warnings, or precautions to take.
-
- The lot number, serial number, or an equivalent symbol.
-
- The UDI carrier directed in Part C of Annex VI of the MDR. Each active implantable device must have its own unique serial number, while other implantable devices only require a serial or lot number.
- An explicit indication of the time limit for safely using or implanting the device, expressed at least in terms of year and month.
- The date of manufacture, which may be included as part of the lot number or serial number.
- There should be an indication of any special storage and/or handling condition.
- For sterile medical devices, labels should additionally include information/symbols regarding the sterilization method, the symbol for sterile, date of validity, and a description of the sterile barrier system.
- Sterile-related details allow manufacturers to mitigate specific risks with aseptic presentation and comply with new legal requirements under EU MDR 2017/745.
- If no expiration date is indicated on the label, the date of manufacture must be present. However, this date of manufacture might be included as a part of the lot or serial number, provided the date is clearly identifiable.
- Information about warnings or precautions that should be made immediately available to the user. The instructions for use may provide more detailed information if this information is kept to a minimum according to Chapter III of Annex I, Section 23.2 EU MDR. Keep in mind that it is up to the manufacturer which warning needs to be included.
- For single-use medical devices, the label should indicate that the device is intended for single use. Additionally, if the single-use device has been reprocessed, there should be an indication of the number of reprocessing cycles already performed, and a clear indication of a limitation on the number of reprocessing cycles.
- If the device is intended for custom-made or clinical investigation only, the label should state “custom-made device” or “exclusively for clinical investigation.”
- The UDI carrier directed in Part C of Annex VI of the MDR. Each active implantable device must have its own unique serial number, while other implantable devices only require a serial or lot number.
3. Labeling under ISO
Besides US and EU requirements, there are several ISO standards for labeling requirements. Check our downloadable guide for an easy overview of the current labeling requirements outlined in ISO standards.
3.1. Information Supplied by the Manufacturer
The 2021 version of ISO 20417 specifies the requirements for information supplied by the manufacturer. It covers the general requirements for identification and labels on medical devices or accessories as well as packaging, marking, and accompanying information and it also covers labels, packaging, marking, instructions for use, and the medical device’s technical description.
If you are a manufacturer of an IVD product, refer to ISO 18113-1 for the terms, definitions, and general requirements of labeling information for IVDs.
3.2. Basic Safety and Essential Performance
If a medical device is designed as medical electrical equipment, the following standards deal with the medical device’s basic safety and essential performance. Let’s have a look at some of these standards:
ISO 80601-2 Series provides basic safety and essential performance requirements for medical electrical equipment. Each Subpart of these standard series contains different specific medical electrical equipment. For example:
-
-
- Part 2-12 addresses critical care ventilators.
- Part 2-56 addresses clinical thermometers for body temperature measurement.
- Part 2-74 addresses respiratory humidifying equipment.
-
IEC 60601-1 Series provides basic safety and essential performance requirements for medical electrical equipment. Each Subpart of these standard series contains different specific medical electrical equipment. For example:
-
-
- Part 1 provides general requirements for basic safety and essential performance.
- Part 1-8 provides general requirements, tests, and guidance for alarm systems in medical electrical equipment and medical electrical systems.
- Part 1-12 provides requirements for medical electrical equipment and medical electrical systems used in the emergency medical services environment.
- Part 2-31 provides particular requirements for the basic safety and essential performance of external cardiac pacemakers with an internal power source.
-
3.3. Use of Symbols
You can use symbols in your label design. Manufacturers mostly prefer symbols over writing because they save space and avoid language barriers. You can use symbols that are defined under an ISO standard or design your own symbol. The key point in designing a new symbol is explicitly defining each symbol wherever possible.
The current 2021 version of ISO 15223 outlines general requirements for symbols that are used with information supplied by the manufacturer. With this updated version, new symbols were added to cover new EU MDR requirements. Part 2 of ISO 15223 was issued in 2010 and outlines symbol development, selection, and validation.
It is important that internationally standardized symbols be used by medical device companies that market their products in several countries with different language requirements for medical device labels so that device safety is not compromised. These standards define symbols to convey essential information for the safe and effective use of medical devices by all users and healthcare professionals across borders and cultural boundaries.
Most Common Symbols Used in Medical Device Labeling