

Quality management is the foundation of medical device manufacturing, as it interlinks with every aspect of the process, from design and development to post-market surveillance. Quality management systems (QMS) are deeply entrenched in the industry as a whole, and you can judge a manufacturer's success on the quality of their QMS.
Even items considered separate from the QMS, such as the technical file, are connected to and impacted by quality management.
How, you ask? Let's get to it!
What is a Medical Device Technical File?
The technical file is a repository of technical information about a medical device's design, development, manufacturing, and testing. It summarizes all the necessary information to evaluate the safety, efficacy, and quality of the medical device and ensure it works as intended.
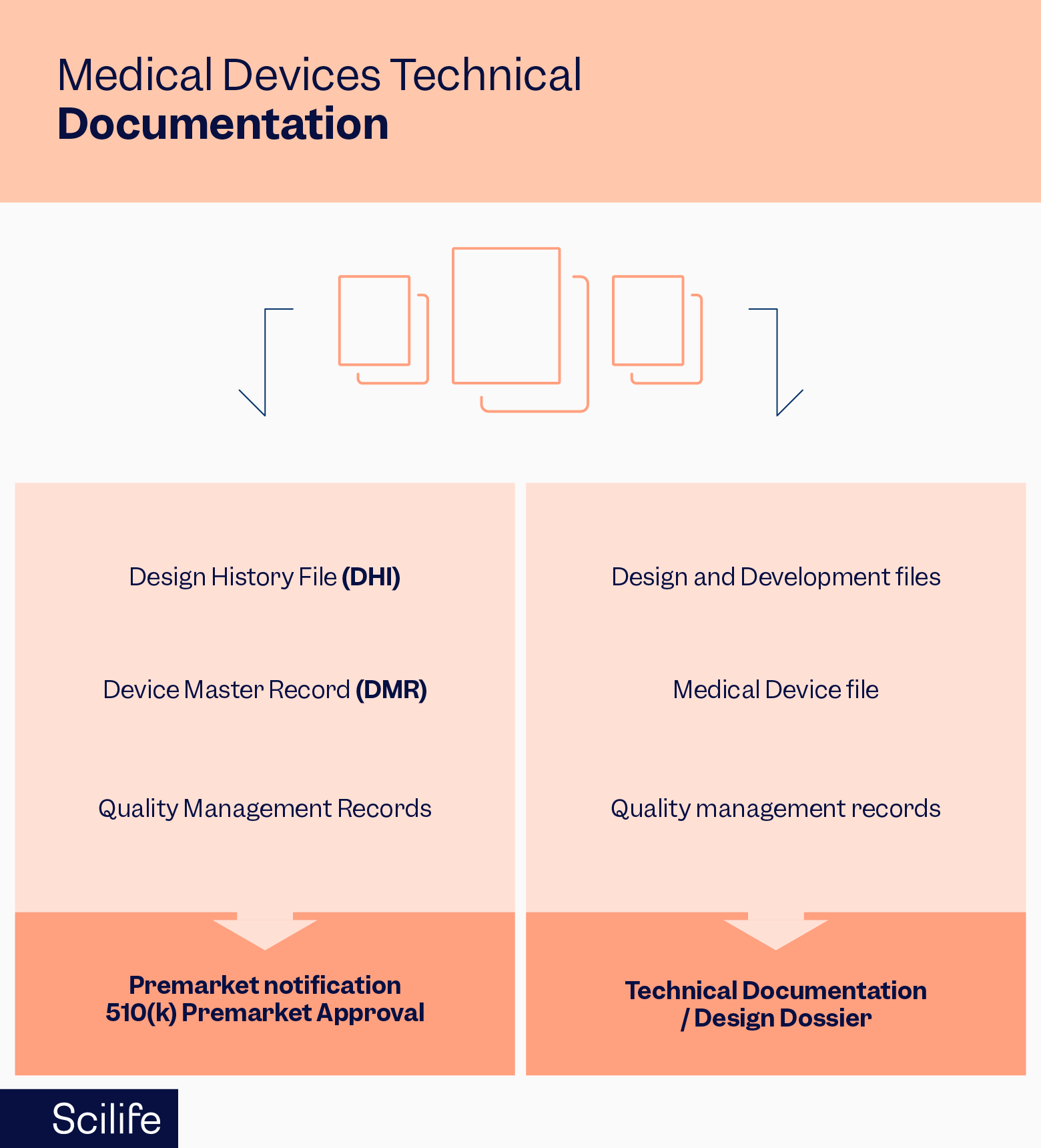
In the EU, the technical file is the dossier submitted to the competent authority. The equivalent in the FDA would be the 510(k) and other regulatory submissions. It is important to note that the design dossier is not the same as a technical file – it is used only for higher-risk devices, much like the US Pre-Market Authorization (PMA).
When is it required, and why is it important?
The technical file is pivotal in the regulatory approval process, whether you seek approval with the Food and Drug Administration (FDA) in the United States or the European Medicines Agency (EMA) in the European Union. The technical file is submitted to the competent authority before market approval. In the EU, it is part of the European Medical Device Regulation (MDR, 2017/745) requirements and thus obligatory for CE marking. Medical devices above Class I self-certified will undergo a notified body technical file review during conformity assessment. The technical documentation in the EU should be updated for ten years after market approval.
While the technical file is essential for conformity assessment and subsequent market approval, it also plays a part in ISO 13485 audits and inspections by the FDA. It also acts as a reference document for regulatory audits and inspections, allowing authorities to assess device conformity.
Content of the Technical File
The technical file in the EU should contain:
- Description and specifications of your medical device
- References to an overview of previous and similar generations of the device
- Labeling and packaging information
- Instructions for use in official EU languages
- Information on the manufacturing and design sites
- Pre-clinical and clinical data
- Post-market surveillance
- Declaration of conformity

In the FDA, the technical file consists of three documents:
Most technical files follow the Summary Technical Document format, STED.
Any technical documentation should be organized and presented so that any reader unfamiliar with the medical device, such as the notified body or regulatory authority, can easily understand and review the information provided. The information should have the right level of detail to make it comprehensive and complete without adding so much detail it bogs down the file.
Traceability of information throughout the file ensures a clear structure and continuity in the review.
Technical file checklist
You can follow the checklist below to ensure you have the correct documentation. The checklist also provides a general structure for your technical file:
- Device description and specifications: general description of the medical device, technical specifications, product and trade names, UDI, etc.
- Information provided by the manufacturer: instructions for use, including warnings, precautions, intended purpose, etc., and packaging and labeling information, which should include hazard symbols, handling instructions, manufacturing and expiration dates, etc.
- Design and manufacturing information: design schematics, design controls, manufacturing process and manufacturing site information, quality control protocols and procedures, and other details on the design and manufacture of the device.
- General safety and performance requirements (GSPR): a description of the GSPR that applies to your device and how to address the GSPR criteria.
- Verification and validation, including biocompatibility testing and clinical evaluation reports, among others.
- Post-market surveillance, including post-market clinical follow-up.
- Risk assessment: risk management reports and other risk assessments and details on the risk management system throughout the lifecycle of your medical device, compliant with ISO 14971.
- Declaration of conformity: names and signatures of the manufacturer or authorized representative, list of standards and regulatory requirements, etc.
Regulatory framework for the technical file
The Medical Device Directive (MDD, 93/42/EEC) described the technical file. However, the regulation could have been more explicit about the content and requirements for the technical file. The technical file requirements were obligatory and relatively similar to the requirements in the MDR, with a few differences. Mainly, the MDD was vaguer and had less emphasis on clinical evaluation and risk assessment, leaving post-market surveillance entirely out of the equation.
The 2016 version of ISO 13485 introduced the medical device file and described the requirements for the device's technical documents. The requirements include device description, intended purpose, labeling, specifications for the device and its manufacture, packaging and storage, market surveillance, and proof of conformity.
The MDR defines the requirements for the technical documentation required before and after CE-marking (as described above), closely resembling the requirements in ISO 13485. The MDR is much clearer on many points and has made it both more accessible and more complex for medical device manufacturers: more accessible because the requirements have been clarified and more complex because they are much stricter than under the MDD.
The notified bodies have also published guidance on the technical file, such as NB-MED/2.5.1. While not legally binding, the guidance is frequently used by notified bodies during audits and conformity assessments.
The FDA requests their technical file documents (design history file, device master record, and device history record) through 21 CFR 820, which deals with quality system regulations of medical devices.
Conclusion
The technical file is a complex amalgamation of the available technical documentation for a medical device. Compiling the technical file can be complicated as documentation is needed from many departments and personnel. Integrating an eQMS can help avoid unnecessary time and resource waste, as you can link your design control records and quickly get available forms. You will also always be audit ready as an eQMS has robust document control capabilities, ensuring you always have the correct document version, audit trail, and electronic signature. You can also set up reminders to keep your technical file updated!
The technical file is a living document and, as such, requires constant maintenance and upkeep. A robust system managing your technical file is a must, whether analog, digital, or automatic.
Do you want to know more about ISO 13485? Watch this video and get all the details you need!





