
In the life science industry, the race is on to earn the prestigious ISO 13485 certification as soon as possible, since it prepares companies for the future EU MDR audit. If you're in the life science industry and after the ISO 13485 certification, this blog post will guide your first steps.
ISO 13485 calls for a lot of work. Where do you even begin?
The first step towards implementing a solution for the ‘quality of science’ is to know the most critical Procedures and Processes within your organization. Knowing these will help in writing a comprehensive Quality Manual that will establish robust Quality Management Practices within an organization.
If you already have the Quality Manual outline in mind, we’ll help you get everything in order. All you need is our enterprise-ready e-QMS platform to implement the QMS practices compliant with ISO 13485.
And finally, before you dive in, take a few minutes to skim through our QARA guide to medical device quality management systems. It’s packed with clarity on what a compliant QMS really looks like and can save you from costly detours later on.
Let’s move on to explain how Scilife enables its users to earn the ISO Certification through its innovative features.
Feature #1: Document Control
Scilife’s Document Control Module helps you manage your documents in the cloud.
Key benefits include:
- Storing your documents on our highly secure cloud-based platform
- Ability to manage different types of of documents
- Documents can be stored in different folders
- You decide who has access to which documents, by assigning user roles
- Documents can be electronically signed by author, reviewer and approver
Why do you need a Document Control Module for ISO 13485 certification? Well, that’s because ISO 13485 mentions various types of documents that a life science company should maintain for implementation of the Quality Management System (QMS) in the organization. You can store and manage all these documents in the Document Control Module without worrying about manual workflow execution, versioning, backup and storage space. It’s a lot less hassle!
General Requirements mentioned in ISO 13485:2016
The Quality Manual is the first document that is mentioned in the 4.2.1 of ISO 13485:2016 standard. It is a document that:
- Formally stipulates the organizational vision about quality with clear statements on quality policy and quality objectives.
- Decides the hierarchy of documents within a QMS.
Once the quality manual is in place, the other documents related to Procedures, Processes, Work Instruction, Templates, and Forms follow.
Control of documents mentioned in ISO 13485:2016
The 4.2.4. of ISO 13485:2016 states that a documented procedure shall define following controls:

To summarise it all, in a paper-based QMS you need a document library and trained resources to create, edit, store and approve formalized documents. However, there is a smarter way of doing it; by using Scilife’s Document Control Module. Scilife's Document Control module allows users to create, edit, revise, store and push the documents through approval cycles digitally in the cloud.
Feature #2: Record Control
4.2.5 of ISO 13485:2016 that states that, “The organization shall document procedures to define the controls needed for the identification, storage, security and integrity, retrieval, retention time and disposition of records.” If you read carefully, you will see that these requirements are no different than 21 CFR Part 11 requirements. Scilife is fully compliant with 21 CFR Part 11. We support you in achieving this as follows:

Feature #3: Events
Section 5.6 of ISO 13485:2016 requires medical device manufacturers to perform a management review of all the Quality Management processes, and the inputs to the review are mentioned in 5.6.2. The input to management defines activities such as complaint handling processes, reporting to regulatory authorities, monitoring measurement of processes and products. It is essential to record all these events for use as input in the management review process. The Events Module in Scilife helps you document all these events, assign a process cause, and perform a risk assessment to investigate the event's root cause as required by 4.1.2 of ISO 13485.
Feature #4: CAPAs
ISO 13485:2016 mentions corrective action in 8.5.2 for reviewing nonconformities (Events), determining the cause of non-conformities, and eliminating the cause of non-conformities through Corrective or Preventive Actions (CAPAs). Scilife’s CAPAs Module streamlines this effortlessly. Writing CAPAs and Events has never been so easy!
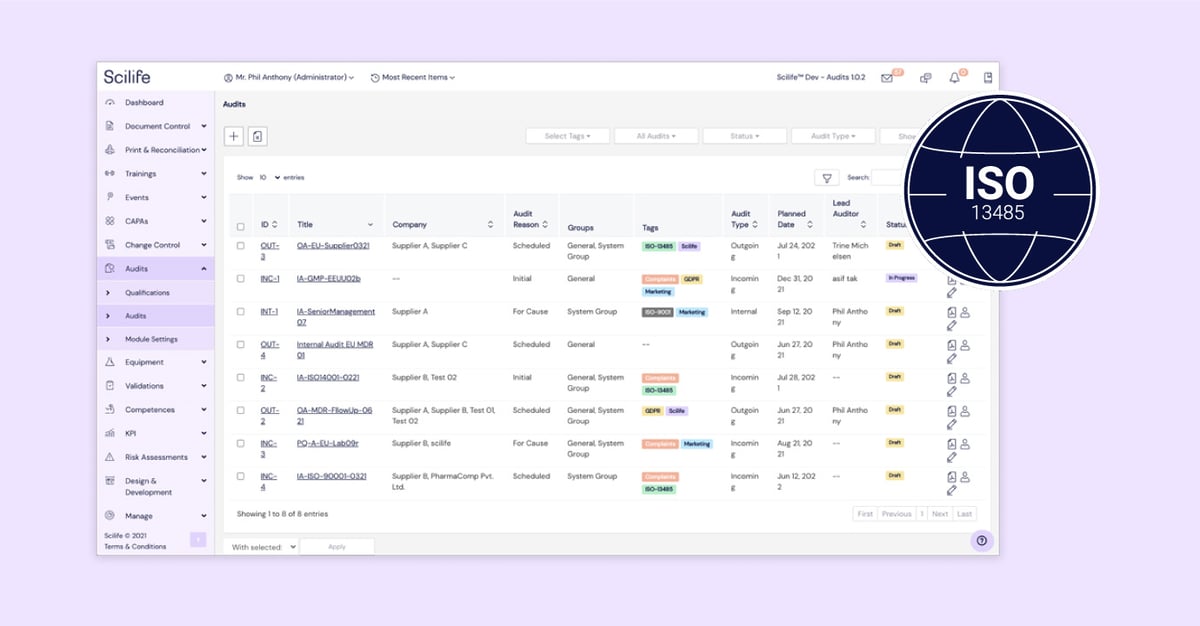
Feature #5: Audit
Vendor or supplier qualification is a crucial requirement mentioned in 7.4.1 of ISO 13485:2016. It states that, “The organization shall document procedures (see 4.2.4) to ensure that purchased product conforms to specified purchasing information. The organization shall establish criteria for the evaluation and selection of suppliers. The criteria shall be:
- based on the supplier’s ability to provide product that meets the organization’s requirements;
- based on the performance of the supplier;
- based on the effect of the purchased product on the quality of the medical device;
- proportionate to the risk associated with the medical device.
The organization shall plan the monitoring and re-evaluation of suppliers. Supplier performance in meeting requirements for the purchased product shall be monitored. The results of the monitoring shall provide an input into the supplier re-evaluation process.
Non-fulfilment of purchasing requirements shall be addressed with the supplier proportionate to the risk associated with the purchased product and compliance with applicable regulatory requirements.
Records of the results of evaluation, selection, monitoring and re-evaluation of supplier capability or performance and any necessary actions arising from these activities shall be maintained (see 4.2.5).
Scilife supports its users in achieving this with the help of the Audit Module. Scilife users can easily leverage the Audit Module to record supplier audit information such as risk assessment and performance in their e-QMS.
Apart from this, the Audit Module is also useful to record internal audit findings for management review.
Conclusion
In a nutshell, Scilife empowers you with all the necessary infrastructure for establishing a fully compliant and streamlined electronic Quality Management System within your organization. The best part, our e-QMS is fully cloud-based and easy to use; requiring no physical storage space, hardware or special workforce!
Curious about how Scilife Smart QMS for Medical Devices can boost your business?
Get in touch!





