

The prime directive of medical device manufacturers is the continued safety of patients through safe and effective medical devices. A critical aspect of medical device safety is ensuring regulatory compliance of any medical device destined for the commercial market. Design controls play a pivotal role in achieving regulatory compliance by providing a systematic framework for managing the design and development process of medical devices and in vitro diagnostics devices.
This article delves into the intricacies of these design controls for medical devices, exploring their purpose, key components, regulatory requirements, and practical tips for implementation.
So, what are design controls, and why do we need them?
Design controls refer to a set of systematic procedures implemented throughout the design and development lifecycle of medical devices. Their primary purpose is to ensure that devices are safe, effective, and meet the intended user needs and regulatory requirements. By incorporating design controls throughout the lifecycle of their medical devices, manufacturers can mitigate risks, maintain quality, and continuously improve their devices.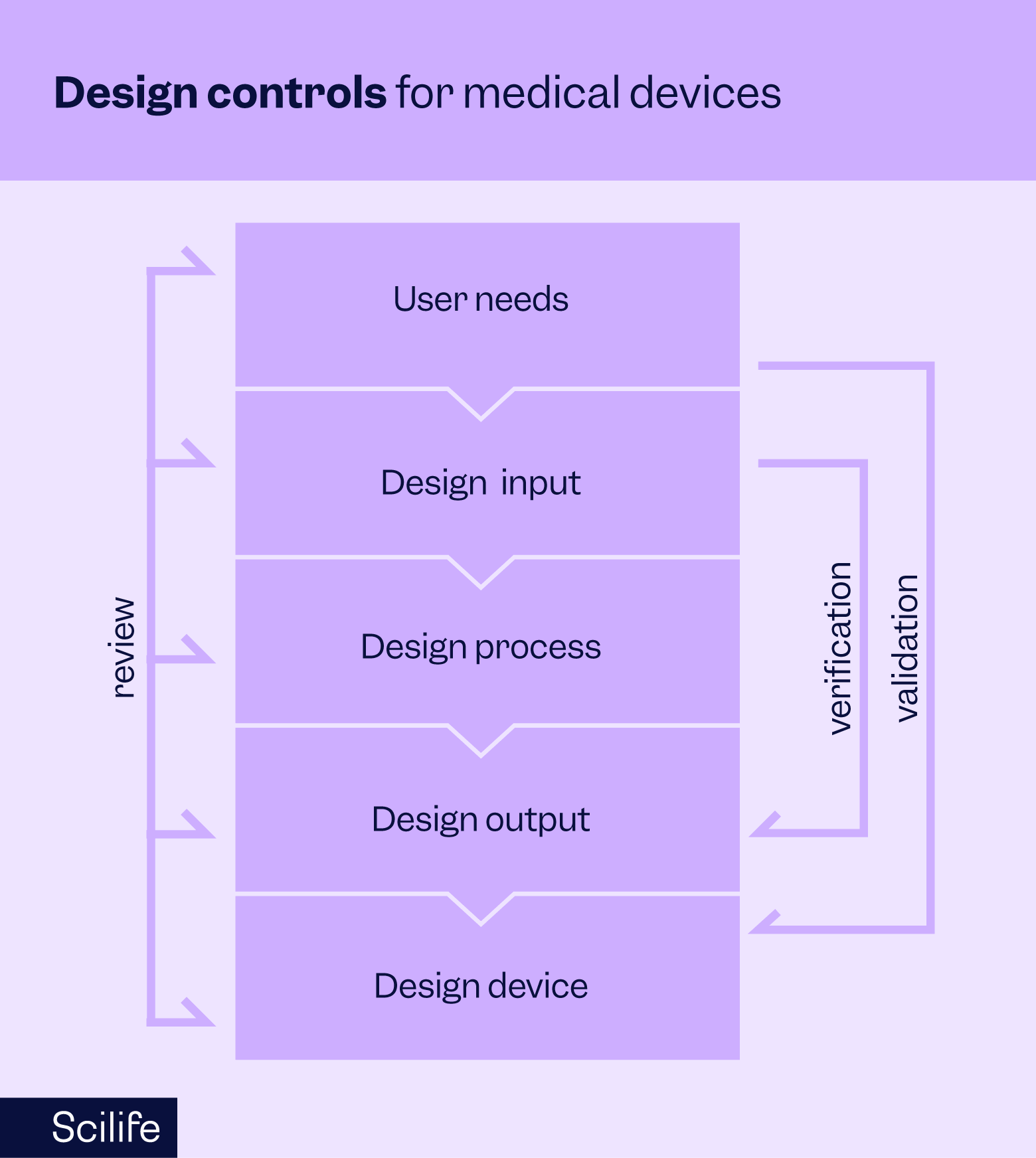
Here's an explanation of each design control:
Design and development planning:
This initial design control phase involves establishing clear objectives, defining the project scope, allocating appropriate (enough) resources, and developing a comprehensive plan to guide the design process. Effective planning ensures alignment with regulatory requirements, timelines, and budget constraints; its importance cannot be overrated.
Design input:
Identifying and documenting the intended use of your medical device, its user needs, and the product requirements that will govern and control the design and development efforts is crucial at this stage. Input from stakeholders, including healthcare professionals and end-users, is precious at this stage and critical to accurately defining these specifications.
Design output:
Generating detailed specifications, drawings, and other documents that define the device design and its components. Clear and comprehensive output documentation is a manufacturing and quality assurance process blueprint.
Design review:
Conduct systematic reviews at various stages of the design process to assess progress, identify issues, and ensure compliance with requirements. Multidisciplinary review teams evaluate design documentation, identifying potential risks and opportunities for improvement.
Design verification:
Performing objective tests and analyses to confirm that the device design meets specified requirements and functions as intended. Verification activities include laboratory testing, simulations, and analysis of design outputs against predetermined criteria.
Design validation:
Demonstrating through objective evidence that the medical device in question meets its user needs and intended use in its intended environment of use. Validation involves clinical evaluations, usability studies, and real-world testing to ensure the device performs safely and effectively.
Design transfer:
Ensuring a smooth transition of the device design from development to production, including documentation, training, and process validation. Transfer activities involve collaboration between design, manufacturing, and quality assurance teams to minimize risks during scale-up.
Design changes:
Implementing controls for managing changes to the device design post-production, including evaluation, documentation, and validation as necessary. Change control processes ensure that modifications are systematically assessed for impact on safety, efficacy, and regulatory compliance.
Design Controls According to FDA 21 CFR 820 vs. ISO 13485
So now that we have the device controls down, how are they presented in the two major quality system regulations?
FDA 21 CFR 820:
The FDA's Quality System Regulation (QSR) outlines specific requirements for design controls in the United States, emphasizing documentation, traceability, and risk management throughout the design process. Compliance with FDA regulations is essential for market approval, quality, and safety. Not to mention, FDA compliance is mandatory for any medical device manufacturer operating in the US.
ISO 13485:
The international standard for quality management systems in the medical device industry guides design control processes, emphasizing a risk-based approach, lifecycle management, and alignment with regulatory requirements worldwide. While not mandatory, ISO 13485 certification is the most frequently used QMS standard in the EU and demonstrates a manufacturer's commitment to quality and regulatory compliance on a global scale.
Tips for solid design controls
Clear documentation: To ensure traceability and compliance, maintain thorough documentation throughout the design process, including design inputs, outputs, reviews, and changes. Document control procedures should facilitate version control, approvals, and access to relevant stakeholders.
Cross-functional collaboration: Foster collaboration among multidisciplinary teams, including engineers, clinicians, regulatory experts, and quality professionals, to incorporate diverse perspectives and expertise. Regular communication and stakeholder engagement enhance transparency and alignment with user needs and regulatory requirements.
- Risk management: Integrate risk management activities into the design process, identifying and mitigating potential hazards and ensuring that residual risks are acceptable. Risk management tools like Failure Mode and Effects Analysis (FMEA) help prioritize risks and implement appropriate controls to minimize adverse events.
- Validation testing: Conduct comprehensive validation testing to confirm that the device meets user needs and performs safely and effectively in real-world conditions. Validation protocols should be well-defined and incorporate relevant test methods, acceptance criteria, and statistical analysis to provide meaningful results.
- Continuous improvement: Embrace a culture of constant improvement, solicit feedback from users, monitor post-market performance, and proactively address any issues or opportunities for enhancement. Post-market surveillance and feedback mechanisms enable manufacturers to identify trends, address emerging risks, and enhance product quality over time.
Learn more by watching our “Medical Devices: Deep dive into Quality Management Systems (QMS)” training series:
How an eQMS can help you with design controls
An electronic Quality Management System (eQMS) can streamline and automate many aspects of design controls to facilitate documentation, collaboration, and compliance. Key features usually include document control, change management, risk management, validation tracking, and audit trails, enabling efficient design process management and reducing the administrative burden on teams. eQMS solutions provide:
- A centralized platform for managing design control activities.
- Ensuring visibility.
- Traceability.
- Real-time access to critical information throughout the product lifecycle.
Design controls are foundational to developing safe, effective, and compliant medical devices. By adhering to rigorous processes and standards, manufacturers can navigate regulatory requirements, mitigate risks, and ultimately deliver products that improve patient outcomes and enhance healthcare delivery. Embracing best practices, leveraging technology solutions, and fostering a culture of quality and innovation are essential in achieving success in today's dynamic medical device industry.
Discover how Scilife smart QMS for Medical Devices can help you easily stay fully compliant.





