The In Vitro Diagnostic Regulation (IVDR) has ushered in a new era of stringent requirements for manufacturers of in vitro diagnostic (IVD) medical devices. Compliance with the regulation is imperative to ensure the safety and efficacy of IVD products in the European market, not to mention mandatory for IVD manufacturers wanting to market their devices in the EU.
Central to IVDR compliance is the establishment and maintenance of a robust Quality Management System (QMS). This guide aims to elucidate the regulatory requirements for IVD devices, provide insights into ensuring QMS compliance, delve into risk management strategies, discuss post-market surveillance obligations, and offer tips and recommendations to achieve full compliance.
Regulatory Requirements for IVDs in EU
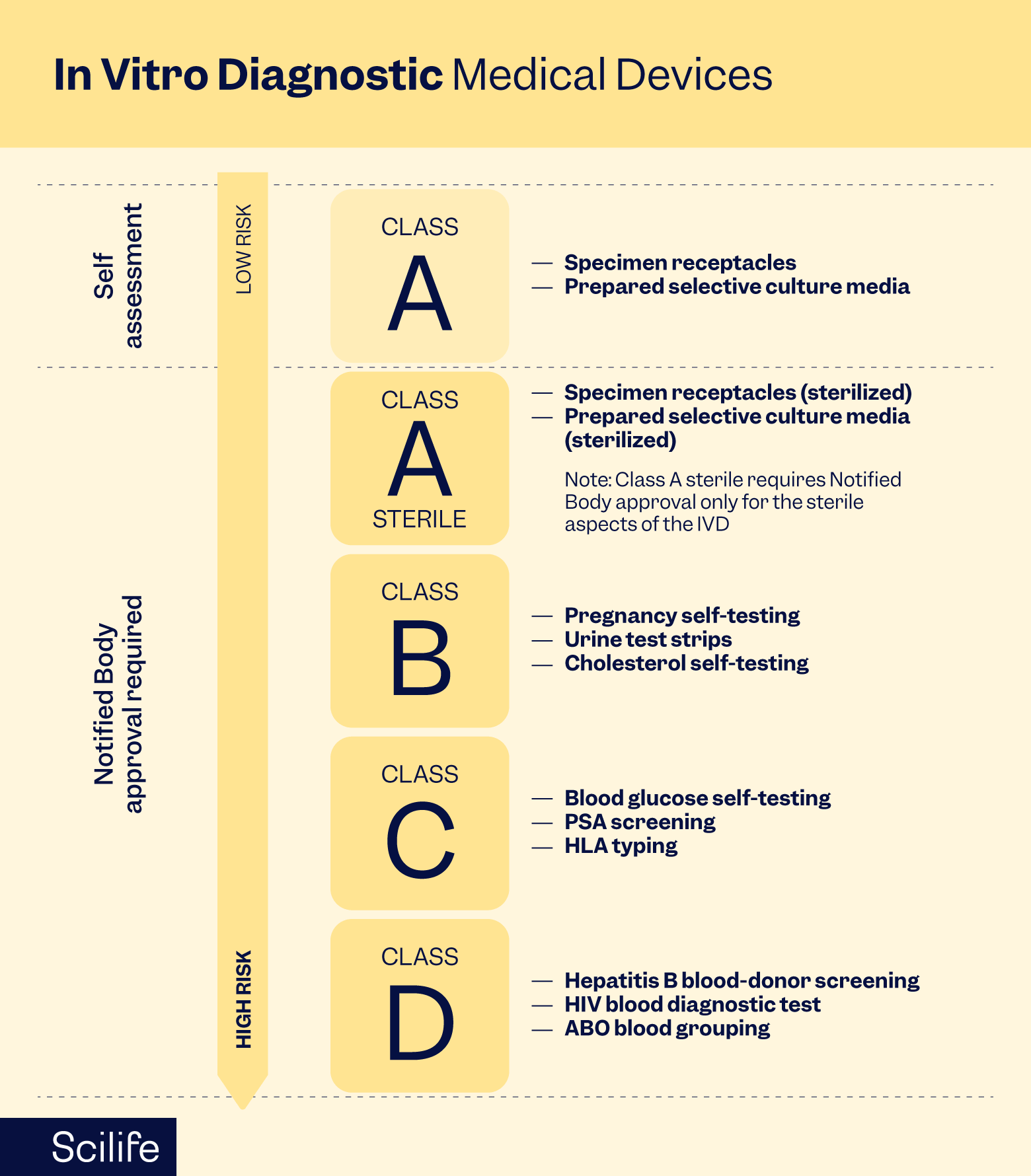
Under the IVDR, IVDs are categorized based on the risks they pose to patients, healthcare professionals, and public health. The regulation classifies devices into Classes A, B, C, and D, with increasing levels of scrutiny and requirements corresponding to the risk class. Manufacturers must adhere to rigorous standards for design, manufacturing, clinical performance evaluation, and post-market surveillance.
How to Ensure Your QMS Compliance
Establishing a QMS tailored to IVDR requirements is critical for compliance. It involves defining organizational roles and responsibilities, documenting procedures, implementing risk management processes, and ensuring traceability throughout the device lifecycle.
Key components of an effective QMS include:
- Document Control: Maintain accurate records of procedures, work instructions, and technical documentation to demonstrate conformity with IVDR requirements.
- Risk Management: Implement a systematic approach to identify, assess, and mitigate risks associated with the design, manufacture, and use of IVD devices.
- Supplier Management: Ensure that suppliers and subcontractors adhere to applicable regulatory requirements and provide necessary documentation to support the quality and safety of components and materials.
- Corrective and Preventive Actions (CAPA): Establish procedures for investigating non-conformities, implementing corrective actions, and preventing recurrence to continuously improve product quality and safety.
- Training and Competence: Provide ongoing training to personnel involved in the design, manufacture, testing, and distribution of IVD devices to ensure they possess the necessary skills and knowledge to perform their roles effectively.
Risk Management
Risk management plays a pivotal role in ensuring the safety and performance of IVD devices throughout their lifecycle. Manufacturers must conduct comprehensive risk assessments to identify potential hazards, evaluate the severity and probability of harm, and implement measures to mitigate risks to an acceptable level.
Key elements of risk management under the IVDR include:
- Hazard Identification: Identify and characterize potential hazards associated with the device, including biological, chemical, and physical hazards, as well as use errors and software failures.
- Risk Assessment: Evaluate the severity of harm that could result from each identified hazard, considering factors such as the intended use of the device, the characteristics of the target population, and the probability of exposure.
- Risk Control: Implement measures to eliminate or reduce identified risks to an acceptable level, such as design modifications, protective measures, or warnings and precautions.
- Risk Communication: Clearly communicate residual risks to users, healthcare professionals, and regulatory authorities through labeling, instructions for use, and other relevant documentation.
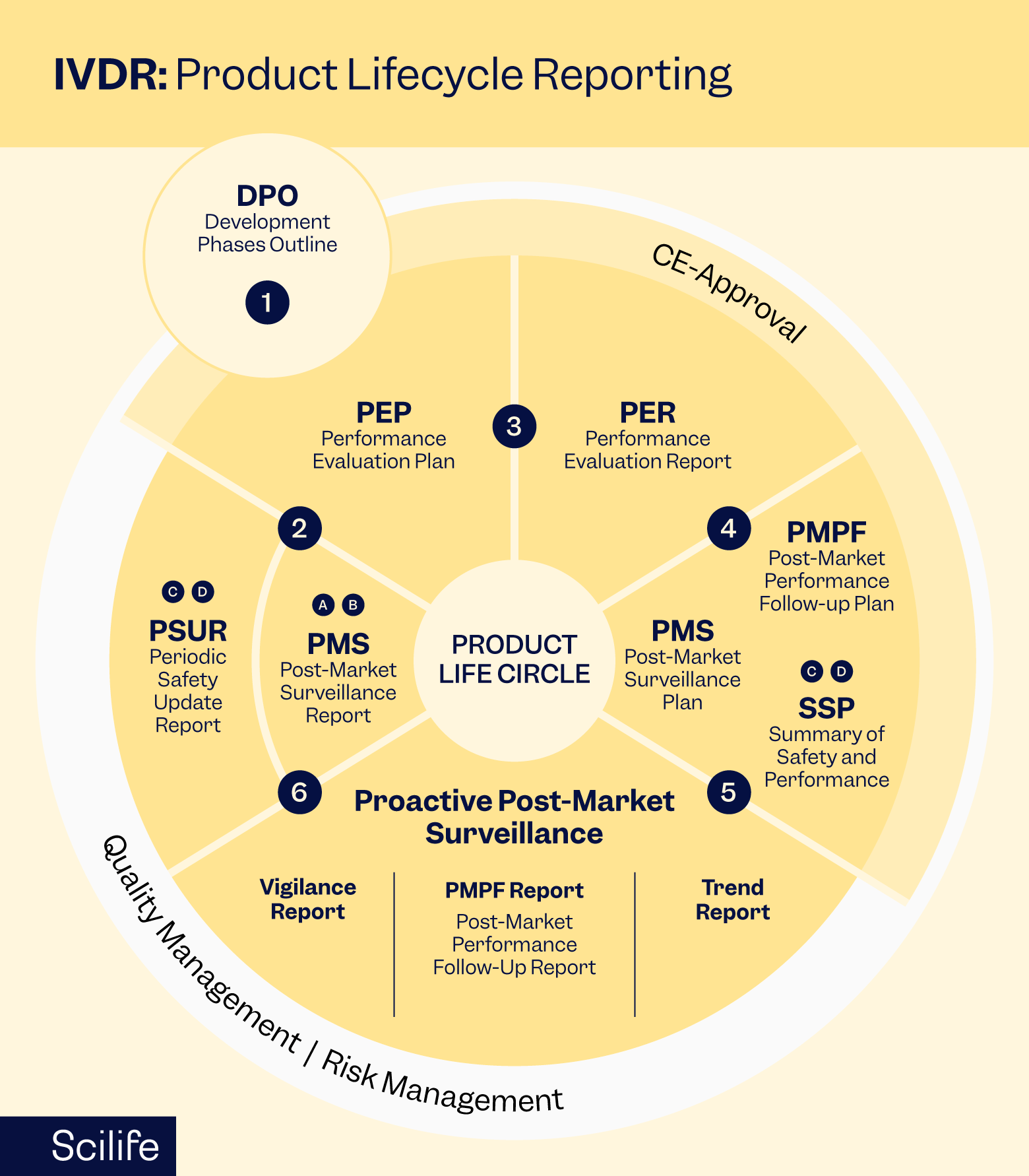
Post-Market Surveillance
Post-market surveillance (PMS) is essential for monitoring the performance and safety of IVD devices once they are placed on the market. Manufacturers are required to establish PMS systems to collect, analyze, and act on data related to the real-world use of their devices.
Key components of PMS include:
- Vigilance Reporting: Promptly report any incidents, including serious adverse events and device malfunctions, to the competent authorities and notify affected users as necessary.
- Periodic Safety Update Reports (PSURs): Prepare and submit PSURs at regular intervals to provide updates on the safety and performance of IVD devices based on post-market surveillance data.
- Post-market clinical follow-up (PMCF): monitor and analyze clinical information to ensure the continued safety and efficacy of your device in clinical settings.
- Trend Analysis: Monitor trends in complaints, adverse events, and other relevant data to identify emerging risks or issues that may require corrective action.
- Continuous Improvement: Use post-market surveillance data to drive continuous improvement initiatives, such as design modifications, labeling updates, or additional training for users.
Tips and Recommendations to Be Fully Compliant
Achieving full compliance with IVDR requirements requires a proactive and comprehensive approach. Here are some tips and recommendations to help you navigate the regulatory landscape effectively:
- Start Early: Begin the transition to IVDR compliance well in advance of the transition deadline to allow sufficient time for implementation and validation of QMS processes.
- Engage with Notified Bodies: Collaborate with notified bodies early in the process to gain insights into their expectations and requirements for IVDR certification.
- Invest in Training: Provide training to key personnel on IVDR requirements, QMS principles, risk management techniques, and other relevant topics to ensure a thorough understanding of compliance obligations.
- Leverage Technology: Explore software solutions and digital tools to streamline QMS processes, facilitate documentation management, and enhance traceability and transparency.
- Stay Informed: Stay abreast of updates and guidance from regulatory authorities, industry associations, and other relevant sources to ensure ongoing compliance with evolving requirements.
Conclusion
Compliance with the IVDR is essential for manufacturers of IVD devices seeking to access the European market. By establishing a robust QMS, implementing effective risk management strategies, conducting rigorous post-market surveillance, and following best practices for compliance, manufacturers can navigate the regulatory landscape with confidence and ensure the safety and efficacy of their products. As the deadline for IVDR implementation approaches, proactive steps taken now can pave the way for successful certification and continued market access for your medical devices in the European Union.