

Medical devices are a vast category of equipment, treatments, and functional aids, ranging from caries detection software to contact lenses, orthopedic implants, respiratory care, and more. Every person will need a medical device at some point or another. We rely on these devices daily and expect them to be safe and perform as the manufacturer intends.
To ensure the safety of medical devices, manufacturers must live up to specific requirements and processes while manufacturing their devices. Quality management standards are implemented by all regulatory authorities in the world in one capacity or another, and they are indispensable aspects of all global regulatory frameworks.
Quality management standards provide standardized and effective requirements for establishing and maintaining a quality management system (QMS) for any manufacturer involved in the medical device industry. Although regulatory authorities historically maintained separate quality management requirements, the FDA now aligns its QMS requirements with ISO 13485:2016 through the QMSR, while the EU and other jurisdictions retain their own frameworks based on ISO 13485
FDA
In the United States, the FDA’s quality standards for medical devices are set out in the Quality Management System Regulation (QMSR), which became effective on February 2, 2026. The QMSR amends 21 CFR Part 820 by incorporating the quality management system requirements of ISO 13485:2016 by reference while maintaining certain FDA-specific requirements to ensure devices are safe, effective, and compliant with the Federal Food, Drug, and Cosmetic Act.
EU
ISO 13485 Quality Management Systems for Medical Devices is the primary quality management standard in the European market. While not mandatory, most medical device manufacturers choose compliance with ISO 13485 to avoid any issues with their quality management systems during manufacturing and audits by the regulatory authority. Likewise, some companies choose compliance with ISO 13485 without going through the certification process to ensure their quality management system is adequate.
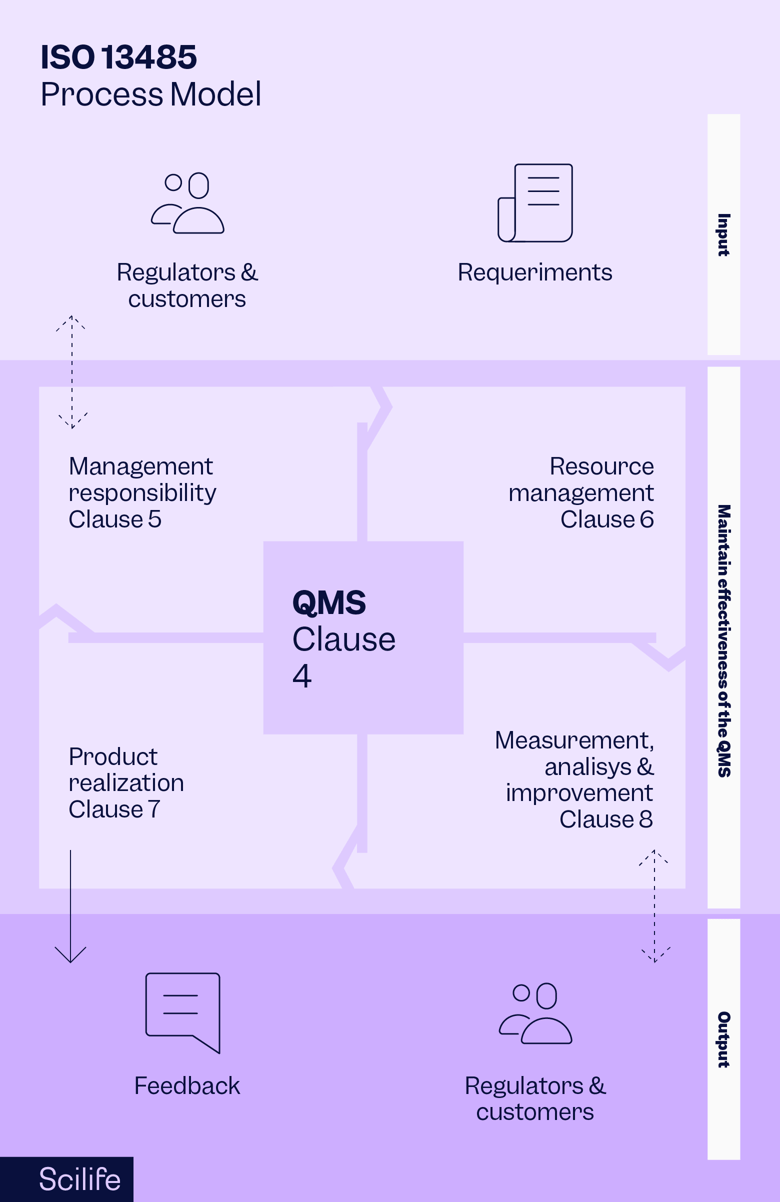
ISO 13485 is designed for use by entities involved in designing, producing, installing, and servicing medical devices and related services. It is also frequently used by other parties during audit processes. The purpose of the standard is to help medical device manufacturers provide medical devices that consistently meet customer and regulatory requirements and maintain patient safety.
There are no exemptions for specific device classes or types within ISO 13485, only aspects of the standard that will be non-applicable for certain devices. For example, if you manufacture a non-sterile device, the procedures associated with sterilization do not apply to you.
The standard is reviewed every five years to check if revisions are necessary to keep up with developments in the industry. The latest version was issued in 2016.
Why is it essential to ensure safety, and how does it benefit the patient and final user?
There's no use for unreliable medical devices. Medical devices should be safe, efficient, and useful. The safety of any product relies on the quality and consistency of the manufacturing process. Almost all device issues originate during manufacturing, which is why there is a real need for control and oversight of medical device manufacturing processes. Pre-market control of medical devices ensures the manufacturer has exhausted every opportunity to improve the safety of their device. Every medical device manufacturer is interested in providing safe products. Still, not every manufacturer fully understands what that requires from their manufacturing facilities and processes – quality standards help manufacturers establish and maintain processes improving their devices as much as possible before they are launched and released to the public.
Some of the measures implemented in quality standards for the protection of patients and users are:
- Protection of employees. Quality management standards help ensure the safety of workers and employees in manufacturing facilities by establishing processes that reduce the risks of falls, electrical accidents, machine hazards, toxic chemicals, temperature accidents, and more.
- Identification and traceability of devices. If a medical device malfunctions or injures a patient, a well-implemented quality management system will help the manufacturer and regulatory authorities trace the device in question. Once the device has been identified, the proper actions to avoid recurrence can be taken, whether it be a recall, a field safety corrective action, or a modification of the instructions for use. Likewise, the QMS processes detail the necessary tasks for each activity, so there is always certainty about the next step.
- Document control. Any quality standard will detail how documents should be drafted, updated, and maintained to ensure there are no gaps in the manufacturers' records. Proper record practices make it possible to trace faulty devices, implement corrective actions, and ensure the entire manufacturing and handling process is optimized for safety and efficiency.
- Labeling and packaging control. The QMS should establish labeling and package control procedures to ensure the patient or user receives adequate information to use the product as intended while minimizing risks as far as possible and that the device arrives at its destination unharmed and in its intended condition.
- Processes for internal and external audits. Any system worth its salt is continuously reviewed and improved. The same goes for quality management systems. They should be tested and checked frequently to ensure they're up to date and fulfilling their purpose. Quality management standards help manufacturers understand the audit process, set themselves up for successful internal audits, and prepare for external audits, for example, by certification bodies.
- Processes for training and capacitating personnel. All employees and personnel involved in manufacturing, distributing, servicing, and storing medical devices should be trained on the QMS procedures applicable to them. A QMS is only as good as the people maintaining it. If personnel are confident of their tasks and responsibilities, the safety and efficacy of the final product are protected.
Medical device quality management system standards are a vital part of the industry. While ISO 13485 is not mandatory in the EU, the EU Medical Device Regulation does require manufacturers to establish and maintain a quality management system.
Because the standards must apply to such a wide variety of devices, both ISO 13485 and 21 CFR 820 are vague and do not describe production in detail. Instead, they provide guidelines for establishing and maintaining procedures that can apply to many different devices and manufacturing abilities.
Likewise, the manufacturer must use their best judgment when determining which parts of the standards to apply to their medical device and specific operation.
Some lower-risk devices remain exempt from certain QMSR requirements under FDA regulations, similar to past exemptions from CGMP obligations. While the specifics of exemptions are listed in relevant sections of the Code of Federal Regulations and can vary by device class, ISO 13485 only allows manufacturers to exclude sections that are not applicable.
Check out how a Smart QMS for Medical Devices can help you follow all key Quality standards.





