
The requirements of life sciences companies (including in the pharma and medical device space) inevitably change over time. Sometimes, a change is due to regulatory requirements; sometimes, customer needs change. Keep reading if you want to learn more about what change is, how to handle change, and what steps you need to take for a successful change control process.
What is Change Control in Pharma and How is it Managed?
Change control management is the process of managing change to ensure that a proposal for a change is properly evaluated, planned, and – if approved – executed and fully implemented. Change control processes are frequently used in the life sciences. With change control management, you will reduce the possibility of unnecessary changes and disruption in the availability of products and back-out activities. Changes affect products, processes, facilities, suppliers, and the source that demands the change, i.e., regulations and customers.
What Is the Purpose of Change Control?
The change control process is mainly based on detailed records with proper traceability through the quality management system (QMS). To establish a living change control management system, you should implement a strong tracing system between each item of change control from several steps. Once you’ve implemented a change control management system, you can see how it changes when an item is updated. You’ll also need to maintain change control records chronologically.
Regulatory Requirements of Change Control Management
Different standards and regulations have outlined the requirements of the change control management process within your QMS.
FDA Regulation
Design change is covered under 21 CFR 820.30:
| “Each manufacturer shall establish and maintain procedures for the identification, documentation, validation or where appropriate verification, review, and approval of design changes before their implementation.” |
Document changes are covered under CFR 820.40, which outlines the requirements for change management in terms of document control:
|
“Changes to documents shall be reviewed and approved by an individual(s) in the same function or organization that performed the original review and approval unless specifically designated otherwise. Approved changes shall be communicated to the appropriate personnel in a timely manner. Each manufacturer shall maintain records of changes to documents. Change records shall include a description of the change, identification of the affected documents, the signature of the approving individual(s), the approval date, and when the change becomes effective.” |
There are also clauses related to FDA’s cGMP for Finished Pharmaceuticals, which provide information about change management under CFR 21 Part 211:
- CFR 21 Part 211.22 - Responsibilities of quality control unit.
- CFR 21 Part 211.100 - Written procedures; deviations.
- CFR 21 Part 211.160 - General requirements.
EU GMP Guidelines
The EU GMP for Medicinal Products for Human and Veterinary Use specify principles and regulations for ensuring standardization in human and veterinary medicines in the European Union. The EU GMP Guidelines are divided into several Annexes describing different processes. For example, Eudralex Volume 4 Annex 15 describes the Qualification and Validation principles. According to Annex 15, change control is defined as follows:
| “Change Control is a formal system by which qualified representatives of appropriate disciplines review proposed or actual changes that might affect the validated status of facilities, systems, equipment, or processes. The intent is to determine the need for action to ensure and document that the system is maintained in a validated state.” |
- CFR 21 Part 211.22 - Responsibilities of quality control unit.
- CFR 21 Part 211.100 - Written procedures; deviations.
- CFR 21 Part 211.160 - General requirements.
EU GMP Guidelines
The EU GMP for Medicinal Products for Human and Veterinary Use specify principles and regulations for ensuring standardization in human and veterinary medicines in the European Union. The EU GMP Guidelines are divided into several Annexes describing different processes. For example, Eudralex Volume 4 Annex 15 describes the Qualification and Validation principles. According to Annex 15, change control is defined as follows:
| “Change Control is a formal system by which qualified representatives of appropriate disciplines review proposed or actual changes that might affect the validated status of facilities, systems, equipment, or processes. The intent is to determine the need for action to ensure and document that the system is maintained in a validated state.” |
Change Control requirements are detailed under Clause 11 of Annex 15.
Chapter 5 of EU GMP describes the guidelines for production, and Clause 5.25 includes guidelines for the change control process:
| “5.25. Significant modifications to the manufacturing process, including any kind of change in equipment or materials, which might affect product quality or the reproducibility of the process, should be validated.” |
ISO 13485:2016
In line with change control requirements of the FDA, ISO 13485:2016 details the terms of change control and design change management under two Clauses. Both Clauses have similar requirements and demand detailed documentation and traceability of every change within a QMS:
Clause 4.1.4
Describes how to manage process changes within your QMS and comply with regulatory change control requirements.
Clause 4.2.4
Describes how to manage document changes within your QMS and comply with regulatory change control requirements.
Roles and Responsibility
There are several roles in conducting a proper change control process. Let’s look at the main roles and their responsibilities through change control.
Change Initiator
The Change Initiator is responsible for and has authority for the following:
- Collecting accurate information to effectively initiate a proposed change, and
- Beginning the initiation process by creating a change record.
For some cases depending on the change itself or the structure of the organization, the Change Initiator may be the Change Owner.
Change Owner (CO)
The Change Owner (CO) has the overall responsibility for the proposed change. The CO is a qualified person to explain the merits of the change and guide the change through the process. They are further responsible for assuring communication of the change package, the reason the change is needed, any mandatory timelines, and for ensuring that the documented system product requirements and/or processes are updated. The CO is also responsible for tracking the change package.
Change Control Board (CCB)
In larger organizations, there might be a Change Control Board (CCB) to manage changes at the entity, project, business, division, or cross-divisional level. The CCB is responsible for:
- Reviewing the technical and business impact elements of a proposed change,
- Defining priorities,
- Dispositioning change packages,
- Ensuring resources are available and adequate, and
- Monitoring changes through the steps of the change process.
Design Owner Organization (DOO)
The Design Owner Organization (DOO) ensures the appropriate management and execution of the product/process changes. The DDO is tasked to:
- Provide guidance and resources to ensure the required change activities are conducted,
- Ensure the documentation resulting from these activities is compliant with the applicable change control procedures,
- Define and assign program/project team member(s) and the majority of the product support team or life cycle management team members,
- Ensure the proper analysis of field data, documentation of proposed initiatives, and compliance with the applicable design/development and product control procedure, and
- Ensure that personnel associated with the management and execution of the change are qualified.
Product Design Owner (PDO)
The Product Design Owner (PDO) is responsible for reviewing risk management documentation and the applicable Design History File (DHF). They also:
- Ensure proper analysis of field data, technical documentation of proposed initiatives, and compliance with the applicable design/development and change control procedures relative to their product, and
- Approve moderate to major product or process changes.
The responsibility of the PDO can be formally delegated to the SME if approved by the DOO.
Quality Representative
The Quality Representative assesses the quality impact of the proposed change, its severity, and the classification of change while providing quality oversight for the execution of all change activities.
Subject Matter Expert (SME)
The Subject Matter Expert (SME) provides a high level of expertise and understanding of the process and/or product.
Change Control Management Process Overview
For effective change control, a procedure should be implemented through a series of steps, which need to be described in the quality manual. The change control management process can be outlined in the following steps:
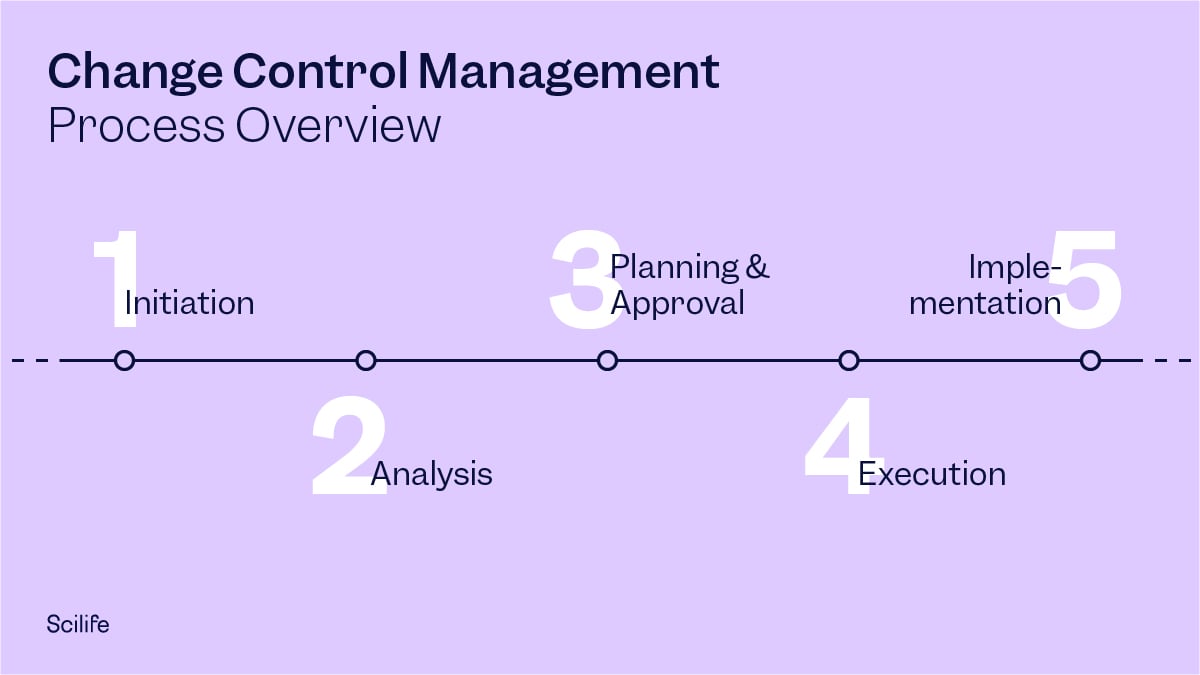
Change Control Process Steps
1. Change Initiation
First, a summary of why the change is needed shall be provided. A change may be needed for various reasons – such as events, regulations, client/third party, or CAPAs.
- Preliminary activities related to evaluation or investigation of the feasibility of a proposed change need to be performed before a change is initiated.
- Change Control Management is initiated after determining the necessity of the change and the scope of the change has been determined.
- If the proposed change is being promoted as an action of a quality system decision (e.g., CAPA), the associated QMS Record shall be linked to the change control record.
A change description should include the following information:
- What triggered the change,
- The scope of the change,
- The current state,
- The proposed state,
- What and who will be impacted by the change, and
- When the change needs to be implemented (if there are mandatory timelines).
1.1 Before Initiating the Change
The CO ensures the change is needed and authorized by:
- Sustaining Products Organization
- Project Management Office
- Change Control Board
1.2 After Initiating the Change
The Change Initiator works with SMEs. Here are some examples of Chane Initiator documents:
- Change title
- Description
- Reason for change
- Change classification
- Products impacted
2. Analysis (Impact Assessment)
2.1 The Severity of the Change
In the initial assessment steps, the severity of the proposed change needs to be determined.
If the proposed change has a potential impact on:
- The safety or effectiveness of the product, including strength, quality, purity, potency, design, material, chemical composition, energy source, or manufacturing process, or
- Modifies the state of validation,that would change product specification requirements, such as:
- The performance of the product,
- Design outputs (form, fit, function), or
- Alters the method of manufacture,
then the proposed change is considered significant. If there is no potential impact, the proposed change is considered insignificant. You must provide a rationale if you decide the proposed change is insignificant. For insignificant changes, skip the following steps, and go directly to the Change Plan:
- Design/DHF Assessment
- Product/Process Impact
- Change Impact Classification
The Quality Representative needs to ensure that the severity of change selection is appropriate. If the severity of change is clear or well understood, the severity should be significant. Insignificant labeling changes may require administrative updates (e.g., a link to the updated Label Specification Number) in the DHF unless these changes only affect the Material Specification.
2.2 The Classification of the Change Impact
You should classify the change based on the results of the Design/DHF/Risk Management File (RMF) and Product/Process Impact assessments. You may classify changes as:
-
Major Changes
Major changes have a substantial impact on the safety or effectiveness of the product, including strength, quality, purity, potency design, material, chemical composition, energy source, or manufacturing process. Major changes can impact the product quality and cause the product to fail in providing its required features. Such changes are irreversible and cannot be corrected, tested, or improved. For major changes, you need to get approved by PDO and RA to be able to continue to the Change Plan.
-
Moderate Changes
Moderate changes have a moderate impact on the safety or effectiveness of the product, including strength, quality, purity, potency design, material, chemical composition, energy source, or manufacturing process. Such changes could affect the process’s reliability; however, they can be corrected, re-tested, or improved. You need to get approved by the PDO and RA to continue to the Change Plan for moderate changes.
-
Minor Changes
Minor changes have a minimal impact on the safety or effectiveness of the product, including strength, quality, purity, potency design, material, chemical composition, energy source, or manufacturing process.
Minor changes impact only a small part of the process without affecting the features of the final product. As a result, the final product compromises all the same quality requirements as before. For minor changes, you need to get approved by the RA to be able to continue to the Change Plan. PDO approval is not required.
2.3 Impact Analysis
The impact analysis shall be performed to determine the impact of the proposed change. Different types of Impact Analysis should be performed depending on the level of the change:
- Quality Impact Analysis
- Regulatory Impact Analysis
- Manufacturing Impact Analysis
All deliverables and change activities identified in the impact assessments shall be reflected in the change plan or have documented justification for why they are omitted. All assessments must be completed before the approval of the change plan.
3. Planning and Approval
- Change plan tasks/deliverables and change implementation approach shall be defined before change plan approval.
- Each task in the change plan(s) shall be categorized as an execution or implementation task, and each plan shall have at least one execution task and one implementation task.
- Product import restriction and product quarantine requirements shall be included as tasks in the change plan to ensure the product manufactured with the change is not distributed to customers before the change implementation approval.
- CO and Quality Representative are the minimum required approvers of the change plan.
- Change plan approval shall be obtained before starting change plan activities.
4. Execution
- Completing execution tasks:
- Review the task and objective evidence requested and target due date.
- Execute the task or assign the task to someone to execute
- Document completion of the task and reference the objective evidence.
- Original records shall reside in the quality system of record.
- Document the completion of each execution task.
- All execution tasks completed, submit for review.
- QA approval:
- Review the execution section of the change plan against tasks completed to ensure tasks were completed to plan and according to requirements.
- Review task records to ensure that objective evidence is provided.
- Complete the review.
- Disposition of the change execution (approve/reject).
5. Implementation
- Complete all implementation tasks:
- Review the task and objective evidence requested and target due date.
- Execute the task or assign the task to someone to execute.
- Document completion of the task and reference the objective evidence.
- Original records shall reside in the quality system of record.
- Document the completion of each implementation task.
- Reference objective evidence that the task is completed.
- QA approval:
- Review the implementation section of the change plan against tasks completed to ensure tasks were completed to plan and according to requirements.
- Review task records to ensure that adequate objective evidence is provided.
- Complete review.
- Disposition of the change execution (approve/reject).
- Close the change.
Benefits of the Change Control Management System
- A Change Control Management system allows you to ensure changes are controlled and coordinated.
- Change Control Management is required for changes to the product, process, facility, and supplier.
- Life Sciences companies use Change Control Management systems to track them thoroughly the entire change control process because of the regulations but also for their patients.
| Integrated | Simplified | Scalable | Measurable |
|
|
|
|
Software for Streamlining Change Control Management
Conventionally, the change control process is executed manually through paper-based systems, which creates a lot of papers that must be appropriately recorded and stored. Too much paperwork only increase the risk of human error and take time to manage. This can result in separated sections, un-signed papers, and record duplication. A better alternative is a software QMS system with a built-in change control management module. It integrates with all quality processes, allows you to link documents easily, and stores everything in a single centralized system. Changes executed using are easily traceable via time-stamped audit trails. Moreover, the system provides secure FDA 21 CFR Part 11 compliant electronic signatures and reporting features allowing for full traceability of your documentation. Document routing, tracking, notifications, and approval all happen automatically, taking control of all integral activities from humans to minimize human error and ensure compliance. By fully utilizing such a system, you can ensure that work instructions and engineering changes can be implemented without anything falling through the cracks.
Determine and Plan Change Requests
Our solution allows you to carefully document the change, including risk and impact assessment, cost estimate, and creating the necessary task pipeline to actually carry out the change. Tasks can be individually assigned to whoever is responsible. If the task requires a document update in the Scilife Document Control feature, the integration between both of these solutions makes it easy to include these documents and enables full traceability within both modules.
Summary
The change management functionality of eQMS software allows users to understand and manage the impact of any change made by your organization and enables users to accurately and easily describe change management processes to internal team members and external auditors. A QMS software solution should be designed with modern best practices that help your team streamline and control the change management process. Whether it be design changes that require updates to design controls and risk matrices, or process changes that impact a myriad of procedures, work instructions, or forms, we’ve got you covered. Scilife’s change management provides users with assurance, knowledge, and real-time visibility of the full impact of change through its automation capabilities while reducing risks to improve team efficiencies and achieve true quality for your products and processes. The ease with which teams can electronically review and approve changes in Part 11 compliant manner can greatly benefit the efficiency of your pre-market design and post-market surveillance activities, as well as facilitate FDA, ISO, and EU MDR audit readiness as you continue to innovate and implement changes to your devices and processes. With a modern, closed-loop quality system, your team will have a single source of truth with full traceability between design controls, risk, documents, procedures, and quality events as changes occur.
Discover how Scilife Change Control Solution can help you establish a clear path for the request, approval, and implementation of modifications, ensuring traceability thanks to the integration with CAPAs and Document Control features.



