Audits and inspections are a part of the quality management lifecycle, they are simply a fact of life in the medical device and pharmaceutical industry. The purpose of an inspection of Quality Management Systems is to ensure your company has established the necessary processes and procedures within your quality management system (QMS), and that those processes and procedures are being followed.
Let’s learn how the FDA CFR Title 21 defines common terms such as audit and inspection.
“Section 820.22 Quality Audit: Each manufacturer shall establish procedures for quality audits and conduct such audits to assure that the quality system is in compliance with the established quality system requirements and to determine the effectiveness of the quality system. Quality audits shall be conducted by individuals who do not have direct responsibility for the matters being audited. Corrective action(s), including a reaudit of deficient matters, shall be taken when necessary. A report of the results of each quality audit, and reaudit(s) where taken, shall be made and such reports shall be reviewed by management having responsibility for the matters audited. The dates and results of quality audits and reaudits shall be documented.”
An inspection, as described in Section 704(a)(1) of the Food Drug & Cosmetic (FD&C) Act [21 U.S.C. 374], involves duly designated officers or employees of the FDA physically entering (at reasonable times and in a reasonable manner), establishments subject to regulation under the Act to determine compliance with applicable FDA requirements (IOM 5.1.1).
“The Food and Drug Administration (FDA) conducts inspections and assessments of regulated facilities to determine a firm's compliance with applicable laws and regulations, such as the Food, Drug, and Cosmetic Act. This typically involves an investigator visiting a firm’s location.”
Today, we’ll delve into details of audit and inspection preparation activities and teach you how to keep your organization inspection-ready at all times.
FDA Expectations of Quality Functions
U.S. Food and Drug Administration (FDA) regulations provide a framework for basic quality requirements that manufacturers must follow. With this framework, the FDA assigns several Quality Unit duties consistent with modern quality systems approaches.
Many regulations refer to QC and QA functions as the quality unit, in which they act together as the governing body for the manufacturing facility, ensuring compliance with good manufacturing practices (GMPs). This means the quality unit ensures the development and implementation of proper standard operating procedures (SOPs) and processes from receipt of raw materials and components to the release of the final manufactured product.
QC usually involves the following:
- assessing the suitability of incoming components, containers, closures, labeling, in-process materials, and the finished products;
- evaluating the performance of the manufacturing process to ensure adherence to proper specifications and limits; and
- determining the acceptability of each batch for release.
QA primarily involves the following:
- reviewing and approval of all procedures related to production and maintenance,
- reviewing associated records, and
- auditing and performing/evaluating trend analyses.
Types of FDA Inspections
A note on terminology: When the FDA comes, it's an inspection, not an audit, although people may casually refer to it as an audit and the FDA inspectors as auditors. Inspections by FDA inspectors will determine whether your QMS complies with 21 CFR Part 820, also known as the Quality System Regulation (QSR).
Routine FDA inspections will occur at least once every two years. Still, any findings at routine inspections may result in compliance follow-up inspections to verify you’ve addressed the problem.
Sometimes, FDA inspectors are unannounced and have the authority to shut down your company on the spot. With the right attitude toward quality and QMS software, there should be no reason to be alarmed when FDA inspectors show up.
The Food and Drug Administration (FDA) performs several types of inspections to ensure that consumers and patients have access to needed products (drugs and medical devices). Inspections fall into four general categories that manufacturers can expect:
#1 Surveillance (also called routine) inspections
This is a standard inspection conducted once every two years for domestic establishments or manufacturers of Class II and Class III devices, and around once every nine years for foreign facilities. If violations are found but corrective actions are immediately taken to address the issues while the FDA inspector is present, this will be noted as a positive indicator of the establishment’s commitment to stay compliant. They are conducted to generally determine or monitor whether a manufacturer is complying with quality manufacturing practices.
#2 Compliance follow-up
If violations are discovered during an inspection, they will be listed on the FDA Form 483. Following that, the FDA will conduct a re-inspection to verify that the issues previously observed have been resolved.
#3 Application-based (Pre-approval, pre-market, or pre-license) inspections
They are conducted, when necessary, as part of the review of an application to market a new product to determine whether it is manufactured in compliance with FDA regulations and to ensure the facility is capable of manufacturing the product consistently and that submitted data are accurate and complete. About 20% of the application reviews are conducted by the agency.
#4 For-cause inspections
They are triggered when there is reason to believe a facility has serious quality/manufacturing problems, to investigate a specific problem/product complaint that has come to the FDA’s attention, or to evaluate corrections that have been made to address previous violations.
The agency also conducts inspections to verify the reliability, integrity, and compliance of clinical and non-clinical research being reviewed in support of pending applications.
Inspections can be a comprehensive review of the entirety of operations at a facility or can be directed, sometimes referred to as a limited inspection, at a particular issue or issues, such as to ensure compliance with recall actions or to follow up on corrective action in the facility.
Challenges: How to prepare for the inspection
Typically, you will receive an announcement at least 5 days before the inspection. The inspection is announced in the majority of cases, and you will have additional prep time before inspectors show up.
However, in the US, the FDA is known to drop in unannounced. This means you should be inspection-ready at all times. So how can you prepare for the audit?
Tips for Preparing for a Successful FDA Inspection
Good preparation helps ensure that your inspection proceeds smoothly and that all issues are covered. However, there may be times when you will not be able to conduct a thorough review of all materials or inspection information before an inspection. Check out the best tips for preparing for a QMS inspection, we listed specifically for you, and make sure you cover them when preparing for your next inspection.
Tip#1: Have answers to all potential questions in advance
It would be useful to do the following activities before an inspection:
- Review all documentation that may be requested by the FDA inspector and ensure that documents are organized and easily accessible.
- The first thing that an inspector starts to review is the company’s history. So you should begin your control by reviewing your company’s history, to include any previous complaints, recalls, personal safety alerts, etc. The purpose of this review is to obtain an overview of your operations and products, understand your compliance history, and be prepared for potential questions with evidence. Conducting a complaint review will help you identify any complaints that require follow-up during your inspection.
- You should also review the establishment file to determine if there are any prior safety issues noted, for example, any documented investigator safety incidents or suggestions for specific personal protective equipment needed before the start of the inspection.
- The endorsement section for the previous inspection, and should be accompanied by a memo to the establishment file.
- If the inspection is being conducted in part or solely as a follow-up to a recall or consumer complaint, you should also plan to review the applicable compliance programs before the start of your inspection. In addition, centers have issued numerous guidance documents for the industry, which you should also become familiar with.
Tip#2: Be Inspection-ready at all times
Maintaining an inspection-ready QMS goes beyond tweaking your standard operating procedures—it's about establishing a culture of quality within your company. Organizations, where everyone understands that quality goes beyond the quality department, are more likely to breeze through audits with no findings.

You can start building a culture of quality and prepare for your next QMS audit by taking these steps.
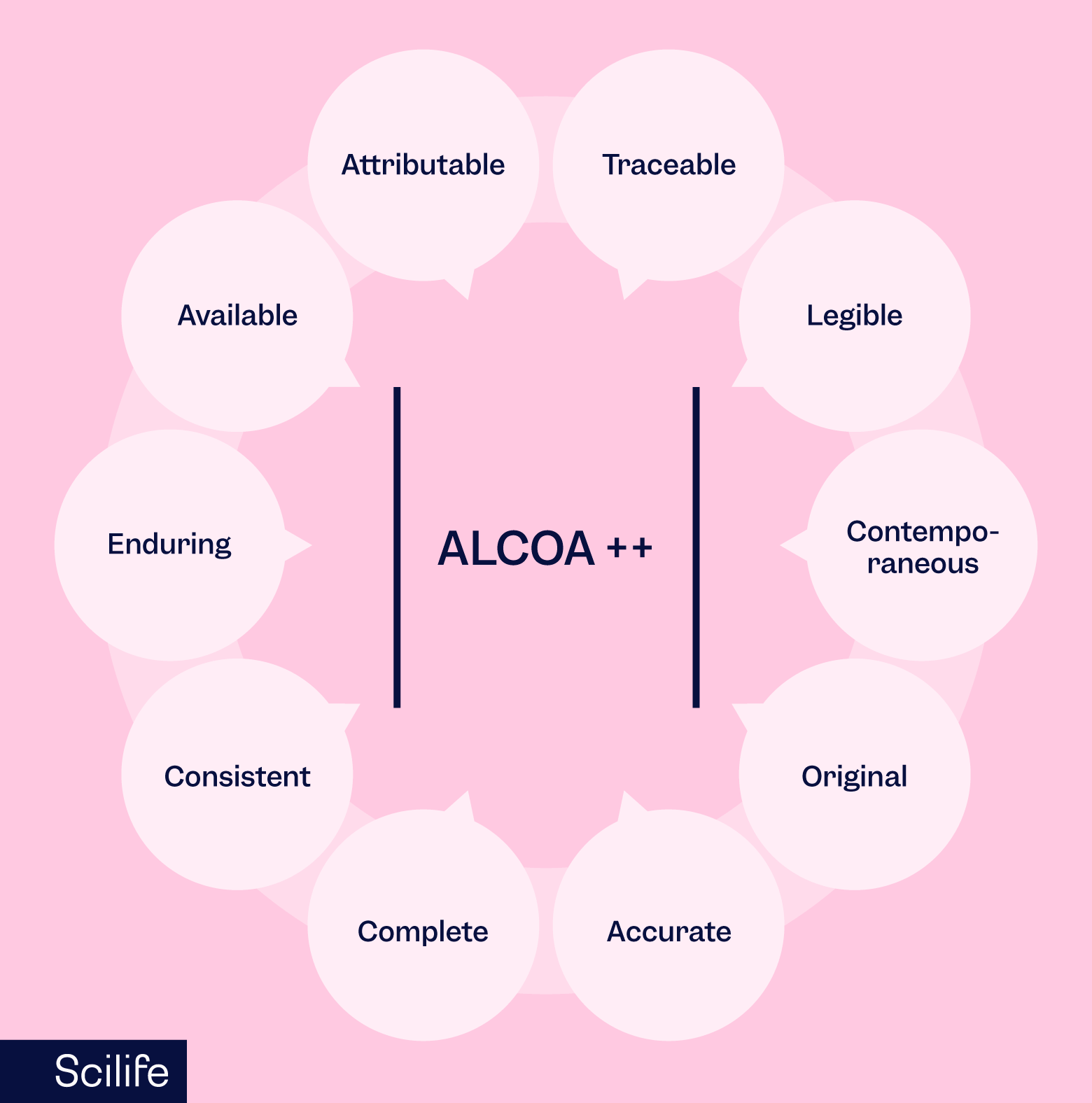
You should not be doing your job from the perspective that it might be audited or inspected”, you do it correctly from the start with the expectations of ALCOA++. The idea that you should:
- document everything as you do the task (and not later)
- file/structure within the flow (and not later)
- organize everything with the expectations of ALCOA++
Tip#3: Know your role
Inspectors usually tend to ask the following questions directly or indirectly, so be prepared to answer:
- Who are you?
- What is your job?
- What is your role in…?
- How were you trained?
- What processes did you follow?
- Were there any concerns?
Each individual in your organization - it could be a worker or a department head - should know the answers to these questions. The first three questions are especially easy to answer, but under stress, people might get tangled up thinking about what their job is and what their specific role in a specific activity is which might be only part of the job which can be tricky to answer sometimes - at least during the execution of these processes. So you and everyone in your organization should think about these questions and prepare to answer in advance.
Tip#4: Know what happened, when, where…
The purpose of an inspection is to seek answers to the following questions:
- What was done?
- Who did it?
- How was it done?
- Why was it done?
- When did it happen?
- Where did it happen?
To respond to these questions, you should be aware of:
- any problems, concerns, or issues that may have happened
- refresh your memory if needed
- where information is stored, how to access it
- who to escalate to
As a quality professional, you can be a quality inspection coordinator or a quality team member in your organization. You should go back and refresh your memory as a good practice especially if you have been inspected in the past. You may need to be aware of problems, the information where it is stored, and how to access it. If the problem happened in the distant past, you might lose access to that system due to the long time that has passed. Therefore, this activity would help you to check whether you need to get new login credentials.
Tip#5: Site preparation before the audit

Typically, people do a lot in the preparation in terms of how to prepare for the interview. Another important thing is to be prepared in terms of logistics.
- Assign responsibilities
- Note taker: somebody who is there, present in the room with interviews. Takes note of all the questions and answers with somebody who is in a secondary room to already highlight as well as inspections
- Quality Coordinator
- Inspector Coordinator
- Define & Prepare room(s)
- A room for the inspectors: they will want to have a separate room to work in
- A secondary room for the team: where you can prepare documents and prepare interviews where you have somebody to support the inspector in case of questions. Ideally, that room is as close as possible so you can see what inspectors are doing and if and when they are leaving their assigned location.
- Prepare documents & materials
Typically this is information related to the quality management system.
-
- One last check on documents and materials that are asked, and needed on the inspection day. If they’re paper-based files, or folders, move them to the secondary room in advance.
- The inspection day before they show up, walk through the premises and remove papers that were left on printers, notices, or notice boards especially if they were outdated, personal notes.
- if you have a reception desk, tell them that there will be inspectors and who they should be calling when they arrive on-site, what happens during an interview, etc.
Tip#6: Tour of the facilities
This is a step that is performed by the inspectors. Our tip for you is to perform a rehearsal of the tour of the facilities. You can act like you’re accompanying the inspectors for the tour of all the locations included in the inspection agenda (if it’s provided in advance). You walk all the rooms, and locations that are in the scope and open each door and closet, check each board, label, etc. make sure everything is controlled and there is nothing present that might be problematic.
What we strongly recommend during the tour of facilities are as follows:
- Following the flow out of a course on the topic that's attended where it's possible and practical
- It is you as a quality lead/inspection coordinator who will guide the inspector. You should keep leading the tour and avoid them taking the lead and go opening doors, and entering areas that you prefer not to show or that are not within scope.
- During the tour, assign a person to keep note of which buildings and rooms they have visited as well as any questions and answers that were provided during the tour.
- Request the inspectors not to take any pictures or videos on premises during inspections due to the organization’s policy. It is sometimes possible that they want to take an image, for example, an image of a setting. Make sure you have the same photograph of the same setting, therefore you will know exactly what they have and what they have seen.
Tip#7: Document and Data Review
During the inspection, the inspector(s) ask you to provide documents and data. Someone (or sometimes more than one person) from quality is responsible for finding and providing documents and data that were asked. The person who is responsible for this and the quality representative or inspection coordinator should pay attention to the following tips:
- If they ask for a random record as an example, provide a completed record and review the record in advance for discrepancies, missing fields, signatures, etc.
- Provide the document or data to the quality representative or audit/inspection coordinator, do not provide records directly to the inspector.
- Maintain a good document flow, and do not delay the provision of documents.
- Preferably, the closest secondary room for the quality team would help you with this.
- Request for documents may come before the inspection, at the opening meeting, during interviews, or ad hoc during document review (ongoing). The quality representative should have clear and fast communication through messages or phone to be able to promptly deliver what was asked.
- Maintain a tracker of documents with their IDs and versions that are provided.
Tip#8: Maintain compliance by conducting effective internal audits
Having a well trained reliable internal audit team can help you to audit all of your internal processes effectively. You would miss valuable opportunities to improve your internal processes and find issues that may result in findings during FDA inspections.
Do you prefer to find and fix the problem yourself rather than let the FDA inspect your facility and issue you a warning letter or Form 483? There seems to be no question about it.
What to expect and how to respond.
Sometimes it feels like the main purpose is to find the answer to “who did it?” Especially, the FDA has the reputation to come in with the idea that there must be a problem and they will find that however, it's not their ultimate purpose. Let’s have a closer look at how to respond in an interview and what behaviors we should pay attention to:
1. Stay calm and professional
Maintain a calm and professional attitude throughout the inspection.
Do NOT argue with or challenge FDA inspectors; focus on providing accurate information.
2. Listen to the question
Listen to the question. If needed, re-phrase in your own words, or ask for clarification
Do NOT interpret, elaborate, or guess.
It's better to seek clarity than to provide incorrect information.
3. Answer the question ONLY
Answer ONLY the question that was asked. Be truthful, factual, and concise.
Do NOT volunteer information beyond what was asked.
Auditors usually use one of the most common interview techniques which is silence. It invites elaboration, and people don't like silence while talking to somebody, they want the conversation to flow. So skilled auditors use silence and you might be inclined to jump into that with more information than they ask for. Instead of providing more information, you can say I.e. “I believe this answered your question”, and then the silence goes back to the auditor.
4. Say if you’re unsure
If you are UNSURE about the answer: it is OK to say. Indicate you will try to find/confirm the correct answer and provide it later. Refer to another colleague
Do NOT invent new things, or try to answer by yourself if the question is not yours to answer.
5. Cooperate and be transparent
Cooperate and be transparent fully with the FDA inspector, providing all requested information. Transparency and openness go a long way in building trust. Contradictory information is always problematic. Therefore, be truthful and complete, referring to documents, and processes as needed for support.
Sometimes the inspector can interview more than one person on the same subject. It means that they ask questions to which they know the answer and they confirm if there is any contradictory information. Therefore, it is so important that everybody needs to be answering the questions correctly also in line with the documentation. In that way, there won't be any conflicts or at least as little as possible between two interviewees. Do NOT mislead with your answer or invent things.
6. Do NOT commit
You can accept the suggestions for improvements.
Do NOT commit to suggestions for improvement at the time of the interview
7. Record observations
Keep a record of the inspector's observations and any issues identified.
This will be crucial for addressing and rectifying any concerns after the inspection.
8. Closing meeting
There will be a closing meeting at the end of the inspection. Inspectors will share all the observations that they have during the entire inspection. Keep the following items in mind during the closing meeting:
- Limit the attendance to key staff only
- Treat FDA inspectors well with hospitality
- If something is not clear, ask for additional clarification
- If you feel certain feedback does not accurately reflect the situation, do attempt to correct it at this stage. do not go into a discussion if it’s strongly needed.
- Make note of observations or ask to receive the draft early
- Confirm where responses to observations should be sent
- Confirm timelines
- Thank the inspector(s)
Handling common audit findings and corrective action plans
Before receipt of the inspection report
- Based on the debrief information at the closing meeting, you may start working on root cause analysis and CAPA(s) may be initiated to manage your time wisely without spending on waiting for the report.
- The report may be different from the debrief at the exit meeting.
Upon receipt of the inspection report
The FDA inspection report shows the final classification into 3 categories for each project area within an inspection:

- No Action Indicated (NAI) which means no objectionable conditions or practices were found during the inspection (or the objectionable conditions found do not justify further regulatory action),
- Voluntary Action Indicated (VAI) which means objectionable conditions or practices were found but the agency is not prepared to take or recommend any administrative or regulatory action, or
- Official Action Indicated (OAI) which means regulatory and/or administrative actions will be recommended.
On the contrary of European audit findings, the FDA works differently. If they have a true concern, they issue Form 483. Otherwise, they will just give you feedback, but not a Form 483. The Form 483 includes findings with categories that we listed above. If it is an Official Action Indicated (OAI), you should tackle it in a formalized response going back to the FDA. The formalization of root cause analysis (RCA) and proper building of causes lies more with items that are seen to significantly impact the regulations or the patients or equality of drugs.
If you received a Form 483, you must perform a thorough Root Cause Analysis (RCA) and based on the outcome(s) of the RCA, issue CAPA(s). The root cause analysis needs to be thorough using multiple methodologies, and based on the RCA you may create CAPA. Sometimes CAPAs seem to come out of nowhere since it's the easiest solution. However, the root cause analysis is the key part of building a good compass.

Solutions
Foundational elements of an inspection-ready QMS.
A facility can ensure an inspection-ready QMS by implementing proper quality control procedures, including clearly written SOPs for assessing the appropriateness of components and closures, monitoring the suitability of the manufacturing environment, and assessing the fitness of manufactured products for release. SOPs must be followed, and QC units must provide accurate documentation of their assessments.
The quality assurance unit must have the authority to police all operations within the facility about the manufactured product. There must be stringent SOPs in place to enable QA teams to review and approve all practices related to manufacturing. QA personnel must have the power to access product quality by reviewing SOPs, maintenance and calibration records, batch production records, and testing records. Finally, QA units must approve or reject all manufactured products for release and must also play a role in auditing all areas of the facility for compliance.
How to implement an FDA-Compliant Quality Document Management System
Your paper-based QMS might be able to meet regulatory requirements, but it doesn’t mean that you have a robust QMS. Having a system to build a more consistent structure for creating procedures, from which you can provide more effective training. Let’s learn some tips for building a consistent structure for procedures:
- Know your purpose. What is the outcome you want?
- Define the scope. When will the procedure be used and when it won’t? What is used as an alternative?
- Define the responsibilities. Who will be responsible and from what? Outline responsibilities and authorities.
- Define all terms and definitions that are used and are specific to the procedure.
- Define your references. How does this procedure interface with others?
- Define the risk. What risks does this procedure address and how does it affect the design of the procedure?
- Define the procedure with a flowchart, and instructions. Outline the process.
- Determine where records are stored and how they are accessed.
- Provide examples for clarity if necessary.
- Test the procedure and evaluate. Update if necessary.
- Determine a revision period
- Define the approval and review.
Scilife’s comprehensive QMS solution helps you stay inspection-ready at all times with Part 11 compliant e-signatures, flexible review and approval workflows, revision control, and more. You’ll be able to provide anything that they ask with the signatures and objective evidence they need as soon as they ask for it. No more chasing down lost or missing records.
Conclusion
In addition to all the tips that we provided you with so far, performing an additional informal meeting with all the participants who were involved in the audit is a very efficient activity to find improvement areas. In that meeting, you should compile all the insights, observations, lessons learned open points, etc. that could have been seen by the inspector but overlooked. It will work wonders! Things that you were not aware of will come to light, and you can compile all your risks, improvements, issues, etc. It also provides a great opportunity to thank all your employees for their effort and engagement in the quality culture. Keep track of internal audit actions and solve as much as possible to be able to get ready better!
Finally, now that you have learned what is written in the report and what was listed in the informal internal meeting, it’s time to act wisely. Treat the FDA inspection as an opportunity for learning and improvement by following this comprehensive FDA inspection checklist and adhering to the tips provided. By doing so, you and your organization will be able to navigate the inspection process with confidence and emerge stronger and more compliant than ever before.
Looking for an FDA inspection-ready QMS? Read more about Scilife smart QMS.